An Overview of Familial Hypercholesterolemia: A Look at Homozygous Familial Hypercholesterolemia, Heterozygous Familial Hypercholesterolemia and LDL Cholesterol Levels
Familial Hypercholesterolemia
Familial Hypercholesterolemia or FH is a hereditary condition characterized by very high levels of total cholesterol and low density lipoprotein cholesterol or LDL-C in the blood since birth. Very high levels of LDL-C, which is also called “bad cholesterol,” causes the formation of atherosclerosis (artery wall thickens) and subsequent cardiovascular diseases in these patients at an early age.
Pattern of Inheritance
For the most part the disease is an autosomal dominant disorder, meaning that a person only needs to inherit the abnormal gene from one parent. When this happens, the condition is called heterozygous familial hypercholesterolemia. In some rare cases, both parents have mutated receptor genes, and the child having inherited both is considered to have homozygous familial hypercholesterolemia.
Genetic Mutation
The defect lies on chromosome 19 where the LDL-C receptor gene is located. It codes for a protein that usually removes LDL from the body’s circulation. However, the mutation produces a protein with reduced binding capacity with LDL-C. In some cases its binding capacity is completely lost. In the presence of these malfunctioning receptor genes, removal of low density lipoprotein cholesterol in the bloodstream is affected, thus elevating its levels. Other genetic defects associated with FH are ApoB mutations which also reduces the receptor gene’s binding capacity, PCSK9 mutations which causes reduction in the number of receptor cells present in the liver, and LDLRAP1 mutations. The function of the protein is still not defined.
Heterozygous Familial Hypercholesterolemia
Heterozygous FH occurs at a rate of 1 in every 500 people worldwide. Children with one parent suffering from FH will have a 50% chance of inheriting the defective gene. Since it runs in families, some relatives of the affected parent may also experience the early development of cardiovascular diseases.
In heterozygous FH patients, the elevation of cholesterol levels start earlier in childhood which results in the premature development of atherosclerosis. With this, cardiovascular diseases could occur at any age depending on the mutation of the LDL receptor gene and other risk factors the patient may have. It is important to detect the disease earlier in order to halt the progress of cardiovascular disease and prevent further complications.
Homozygous Familial Hypercholesterolemia
Homozygous FH is not as common as heterozygous FH. It has a ratio of 1 in every 1,000,000 people worldwide; however, the disease is more severe and often fatal. There is extreme elevation of LDL-C in the blood from birth which results in accelerated atherosclerosis and earlier manifestation of heart problems.
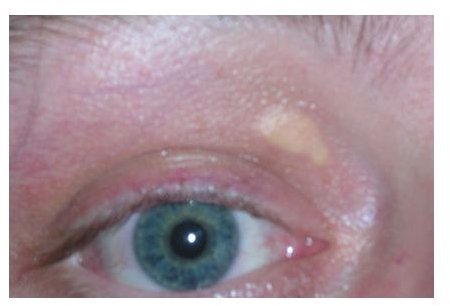
Individuals may also experience cutaneous xanthomas or cholesterol deposits in the hands, elbows, wrist, knees and buttocks. Cardiovascular diseases will usually manifest before the patient reaches puberty, where sudden death is not uncommon. In both types of FH, early detection is essential. When parents or relatives are showing signs of hypercholesterolemia, it is suggested that their siblings and children should undergo further evaluation for earlier detection of the disease. Proper management and treatment could then be given in order to prevent the onset of cardiovascular problems.